
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-precious bimetallic catalysts for selective dehydrogenation of an organic chemical hydride system†
Anaam H.
Al-ShaikhAli
,
Abdesslem
Jedidi
,
Luigi
Cavallo
and
Kazuhiro
Takanabe
*
Division of Physical Sciences and Engineering, KAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), 4700 KAUST, Thuwal 23955-6900, Saudi Arabia. E-mail: kazuhiro.takanabe@kaust.edu.sa
First published on 6th July 2015
Abstract
Methylcyclohexane (MCH)–toluene (TOL) chemical hydride cycles as hydrogen carrier systems are successful with the selective dehydrogenation of MCH to TOL, which has been achieved only using precious Pt-based catalysts. Herein, we report improved selectivity using non-precious metal nickel-based bimetallic catalysts, where the second metal occupies the unselective step sites.
Liquid organic hydride couples, such as methylcyclohexane (MCH)–toluene (TOL), have been potential industrial candidates for efficient and safe hydrogen storage and transport.1 An effective system requires a catalyst with high selectivity for the endothermic dehydrogenation reaction shown in eqn (1).
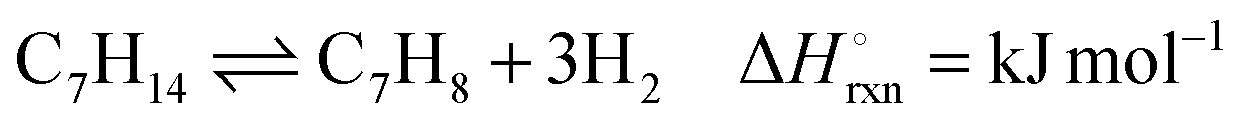 | (1) |
In addition, numerous efforts have been focused on substitution of noble metals with non-noble mono or bi-metallic based catalysts, such as Ni based or Cu based catalysts, for dehydrogenation reactions.8,9 Ni based catalysts are cost-effective but pure Ni based catalysts exhibit a high hydrogenolysis activity (C–C cleavage), which leads to inferior selectivity for dehydrogenation.8 Nevertheless, none of the developed catalysts completely suppress unwanted hydrogenolysis side reactions. Based on the progress towards a hydrogen-based society, the quest for catalysts for liquid organic hydride systems that can technically and economically compete with Pt-based catalysts continues. To tackle this challenge, we report the synthesis and experimental and computational characterization of Ni-based bimetallic catalysts, which exhibits an unprecedented high selectivity for the dehydrogenation of MCH to TOL.
A series of mono- and bi-metallic catalysts were synthesized using a homogenous deposition precipitation method (the detailed synthesis procedure is provided in the ESI†).10Table 1 lists the catalytic results of MCH dehydrogenation at 350 °C using mono-metallic catalysts (Ni, Ag, Zn, Sn, and In) and bi-metallic catalysts (Ni–Ag, Ni–Zn, Ni–Sn, and Ni–In) supported on Al2O3. In agreement with previous results,10 the Pt catalyst used as a reference sample exhibited high activity and high selectivity to TOL (99.9%). Group 11–14 (Ag, Sn, Zn, and In) monometallic catalysts exhibited negligible MCH conversion (<1%), which was most likely due to the lack of catalytically active d-electrons but coupled with the inability to promote the undesired C–C breaking event with Ag, Zn and In (byproducts <4%). Alternatively, Ni resulted in a noticeable conversion (36.2%) but low TOL selectivity (∼67%) with benzene and methane as the major byproducts, indicative of competitive dealkylation of the methyl group on the MCH and/or TOL.
| Catalyst | Conversion (%) | Carbon selectivity (%) | ||
|---|---|---|---|---|
| TOL | Benzene | Methane | ||
| Pt | 85.9 | 99.9 | 0.1 | <0.1 |
| Ni | 36.2 | 66.9 | 25.7 | 5.7 |
| Ag | 0.3 | 93.8 | n.d. | 0.4 |
| Sn | 0.9 | 46.6 | 50.2 | 1.8 |
| Zn | 0.1 | 95.9 | 0.1 | 2.8 |
| In | 0.0 | 91.8 | n.d. | 3.5 |
| Ni–Ag | 14.7 | 71.7 | 21.2 | 6.1 |
| Ni–Sn | 16.0 | 93.2 | 5.2 | 1.4 |
| Ni–Zn | 32.2 | 96.6 | 2.7 | 0.4 |
| Ni–In | 9.8 | 99.5 | 0.4 | 0.1 |
At this point, we wondered whether bimetallic Ni-based catalysts would exhibit improved performance by coupling the good activity of Ni with the high selectivity of the second metal. Notably, the addition of Ag, Sn, Zn, and In (2 wt%) to the Ni (8 wt%) catalyst lowered the MCH conversion compared to the Ni monometallic sample, but, in all of the cases, the selectivity for TOL improved (Table 1). Among the bimetallic catalysts investigated, the Ni–Zn system exhibited high selectivity towards TOL while maintaining high conversion rates, and therefore, this system was selected for more detailed characterization.
Next, we prepared the catalysts with various Ni/Zn ratios. Table 2 lists the metal content determined by inductively coupled plasma (ICP) and metal dispersion measured by a CO pulse adsorption technique. The addition of Zn reduced the metal chemisorption properties per Ni. This result most likely indicates that Zn occupies the surface Ni sites to avoid interaction with the CO molecules based on the lack of chemisorption by the Zn monometallic catalyst, which is consistent with previous results.11
The MCH conversion as a function of time for the Ni, NiZn0.1 and NiZn0.6 samples at 300 °C is shown in Fig. 1a. The Ni and NiZn0.1 catalysts were comparable in their conversion levels even though the CO chemisorption capacity was ∼40% lower with NiZn0.1 than with Ni. NiZn0.6 which has a lower metal dispersion exhibited a relatively lower conversion than these two samples but not to the extent that was expected by the CO chemisorption capacity, which was 6.2 times lower for NiZn0.6 than for Ni. This result suggests that the NiZn bimetallic catalysts exhibit a relatively high turnover frequency based on the CO chemisorption capability. All of the catalysts exhibited relatively stable performance with ∼10% conversion loss after 500 min.
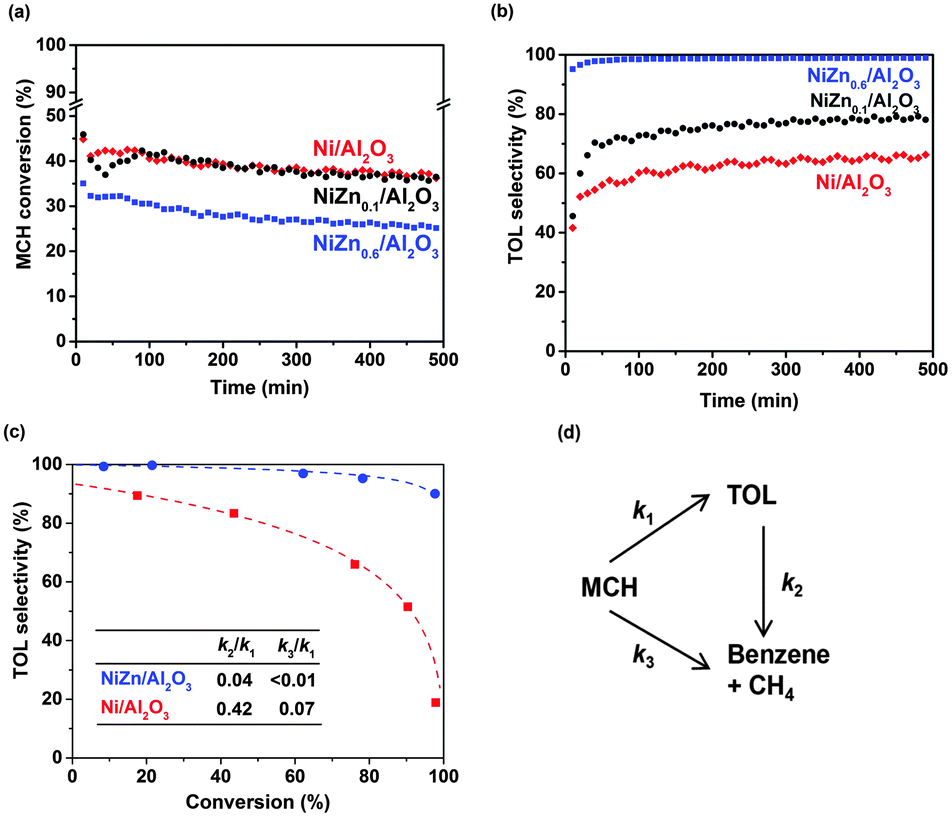 | ||
| Fig. 1 (a) Conversion of MCH and (b) selectivity to TOL as a function of time using Ni/Al2O3, NiZn0.1/Al2O3, and NiZn0.6/Al2O3. 20 mg of catalyst, 400 °C, H2 reduction, 300 °C, 42.86 kPa H2, 1.37 kPa MCH at 20 °C, 4.76 × 10−6 g h ml−1. (c) Selectivity to TOL as a function of MCH conversion for Ni/Al2O3 and NiZn0.6/Al2O3. 5, 20, 100, 200 and 500 mg of catalysts, 400 °C H2 reduction, 300 °C reaction, 50 kPa H2, 2.4 kPa MCH, 2.08 × 10−6–2.08 × 10−4 g h ml−1. (d) A simplified reaction scheme. | ||
Fig. 1b shows the selectivity towards TOL as a function of time under the same conditions as those employed in Fig. 1a. In the beginning, all of the catalysts exhibited improvement in the TOL selectivity, suggesting that reconstruction of the surface active sites occurred during the catalysis. Reduction of metals during the reaction was unlikely for the reason because the reduction temperature (400 °C) was much higher than the reaction temperature (300 °C). In the literature,12 it was proposed that dehydrogenation proceeds on the carbonaceous deposited surface, causing induction period. In some cases, the observed dehydrogenation rate was higher than the clean surface based on the induction period.13 Understanding the phenomena during this induction period requires additional detailed investigation. However, the bimetallic catalysts exhibited obvious improvement in TOL selectivities. The Ni/Al2O3 sample exhibited a low selectivity to TOL (i.e., ∼60%). The addition of 10 mol% Zn to the Ni catalyst resulted in minimal but obvious improvement in the selectivity to ∼75%. When the NiZn0.6/Al2O3 catalyst was employed, the TOL selectivity was substantially improved to 99%.
For better comparison of TOL selectivity at different conversion levels, the space velocity was varied by changing the catalyst loading in the reactor. Fig. 1c shows a comparison of the TOL selectivity as a function of conversion. For the Ni catalyst, the selectivity extrapolated to zero conversion did not reach 100%, suggesting that the hydrogenolysis (C–C cleavage) of MCH to CH4 and cyclohexane, followed by dehydrogenation to TOL, occurs (non-zero value for k3 in the scheme shown in Fig. 1).14 In contrast, for the NiZn0.6 sample, the selectivity to TOL reaches nearly 100% at low conversion, which indicates the complete suppression of MCH to CH4/benzene (hydrogenolysis products: negligible k3) and selective TOL formation (dehydrogenation product). The loss of selectivity using NiZn0.6 may be due to the transformation of TOL to CH4 (0.09%) and benzene (0.56%) under the conditions investigated (secondary conversion of TOL: non-negligible k2). For the Ni-only catalyst, the TOL selectivity decreased to 18.8% at a higher conversion of 97.9%. This poor selectivity originates from large rate constant for the secondary reaction of TOL to benzene and CH4 (k2). In contrast, NiZn0.6 maintains a high selectivity (90.0%) at high conversion (97.7%), which demonstrates minimized contribution for the secondary conversion of TOL and its potential for use as a highly selective catalyst under the same experimental conditions. These kinetics are apparent from the pseudo-first order rate constant in the reaction pathway shown in Fig. 1d. Further reduction of the k2 value is still essential, requiring future study to reduce binding energy of TOL products. The promotion effect due to the addition of Zn to Ni may be due to changes in the electronic/geometric and adsorptive properties, which promotes C–H cleavage rather than C–C cleavage, reported for various other hydrocarbon reactions.15,16
Typical high-resolution transmission electron microscopy (HRTEM) and scanning transmission electron microscopy (STEM) images for the Ni and Ni–Zn0.6 based catalysts after reduction at 400 °C are shown in Fig. 2 and Fig. S1 (ESI†), respectively. The metal nanoparticles correspond to the black dots in the HRTEM mode in all of the samples, and the size of the nanoparticles were determined to be approximately 2–5 nm irrespective of these samples. The introduction of Zn had a minor influence on the size of the metal nanoparticles. Differentiation between the Ni and Zn elements was difficult due to their comparable electron densities. The XRD patterns of the reduced Ni, Zn and Ni–Zn0.6 samples supported on Al2O3 are shown in Fig. S2 (ESI†). Consistent with the small particle size obtained from the HRTEM images (Fig. 2), the metallic (or alloy) peaks were not clearly visible and overlapped with the peaks of the γ-Al2O3 support. For the Zn/Al2O3 catalyst, the minor peaks observed in the XRD patterns suggested the presence of hexagonal ZnO, which was less evident in the NiZn0.6 sample. For NiZn/Al2O3, the XRD pattern does not provide clear evidence for phase identification.
 | ||
| Fig. 2 Representative HRTEM images of (a) Ni/Al2O3 and (b) Ni–Zn0.6/Al2O3 reduced at 400 °C. | ||
Understanding the impact of the catalyst structure on the activity and selectivity of hydrogenolysis vs. dehydrogenation of hydrocarbons is fundamental for the development of improved catalysts. Nørskov and coworkers reported that a NiZn alloy can be a selective catalyst for the hydrogenation of acetylene in the presence of ethylene.16 This claim was based on the hypothesis that ethylene adsorption/hydrogenation can be particularly competitive at steps and other low-coordinated sites on the catalyst surface and these sites could be poisoned by Zn. In addition, the same step and low-coordinated sites were associated with C–C bond breaking.8,17 This scenario suggests that Zn could poison low-coordinated C–C bond breaking sites in our nanoparticle NiZn catalysts. To support this hypothesis, we performed density functional theory (DFT) calculations.
Initially, we modeled a 55 Ni atom cluster with icosahedron and cuboctahedron symmetries (Fig. 3). The icosahedron structure was more stable than the cuboctahedron structure by 3.75 eV,18 and therefore, we focused on the icosahedron structure in the following Zn substitution study. Next, we modeled a series of Ni54Zn clusters where a Zn atom replaced a Ni atom in an icosahedron Ni55 cluster at different positions. On the surface, which corresponds to the second Ni shell, the Zn atom can be placed on a corner or an edge. The geometries with the Zn atom at these two positions possess the same stability (i.e., Zn on the edge is only 0.01 eV higher in energy, see Fig. 3). Taking the geometry with Zn at the corner as a reference at 0 eV, the geometry with the Zn atom in the first coordination shell is 1.63 eV higher in energy, and the geometry with the Zn atom at the center of the cluster is 1.51 eV higher in energy. Overall, these results indicate that the Zn atom prefers to be on the surface rather than in the core of the NPs, and this result was confirmed with cluster sizes of 13, 55 and 147 atoms.
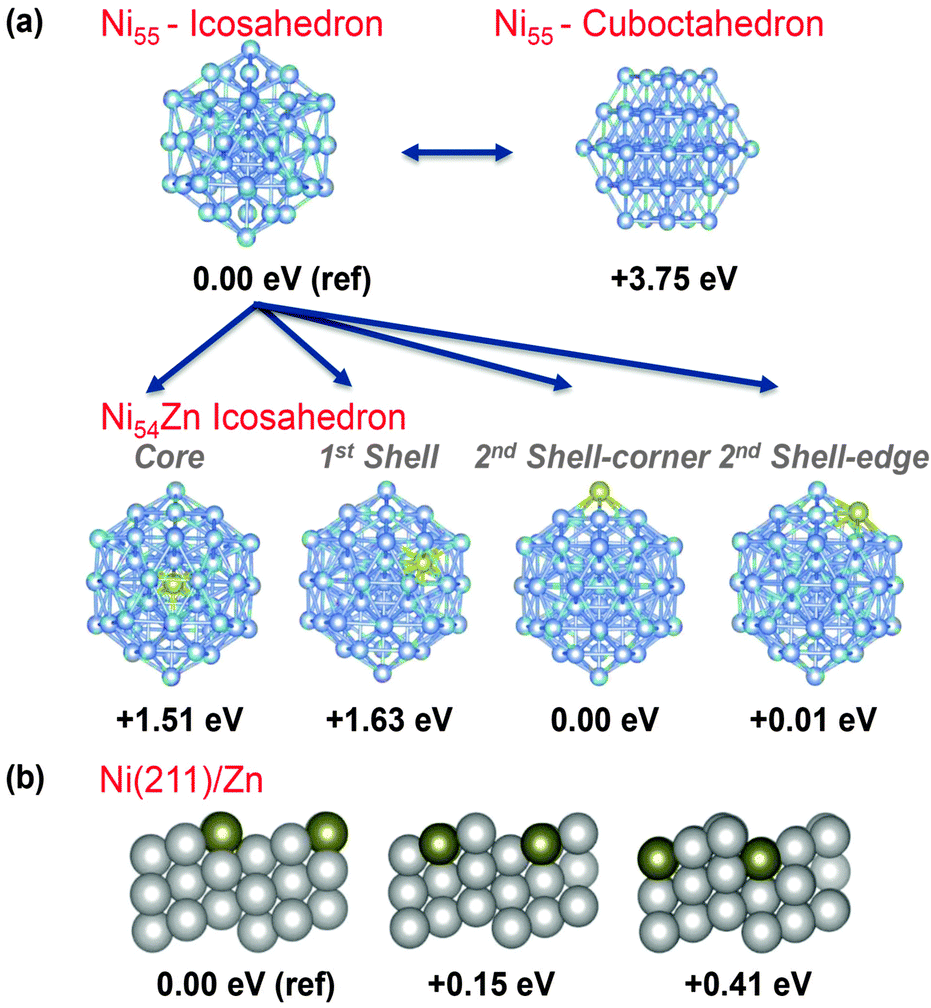 | ||
| Fig. 3 (a) Site preference of Zn replacing one Ni in a Ni55 icosahedron cluster. The energies relative to this cluster are presented. (b) Side views of the three possible geometries of the (211) facet with one Zn atom replacing a Cu atom. The energies relative to the most stable structure (left) are presented. | ||
To further explore the Zn preference for low-coordinated sites, we modeled the effect of replacing one Ni atom with a Zn atom on the (211) facet of Ni because this facet is stepped and contains 3 different types of Ni atoms. The Ni atom types range from highly unsaturated at the ridge of the step to highly saturated at the bottom of the step (Fig. 3b). Consistent with the calculations performed on the NiZn nanoparticles, the geometry with a low-coordinated Zn, such as the ridge of the step, is the most stable, and the geometries with the Zn in the middle or at the bottom of the step were less stable (Fig. 3b). Consistent with the results from previous studies,17 this result further supports the hypothesis that low-coordinated sites are active in C–C breaking and can be poisoned by the preferential substitution of Ni by Zn.
In conclusion, we rationally designed and confirmed that a Ni-based bimetallic catalyst offers an attractive solution for the selective dehydrogenation of MCH to TOL. The NiZn0.6/Al2O3 catalyst exhibited outstanding performance with a slightly reduced conversion coupled with excellent selectivity compared to that of the corresponding Ni/Al2O3 catalyst. The results obtained from DFT calculations suggest that the main role of Zn is poisoning of the low-coordinated sites where C–C breaking preferentially occurs, which improves the selectivity and leaves Ni atoms in the center of the facets of the nanoparticles performing the dehydrogenation. This study provides an economically viable catalyst with good performance as well as a clear route for the design of even more selective catalysts to challenge the established role of expensive Pt-based catalysts.
The research reported in this work was supported by the King Abdullah University of Science and Technology (KAUST). A. H. S. acknowledges Saudi Aramco for financial support. L.C. and A. J. are grateful to the KAUST Supercomputing Laboratory for the resources provided under the project k199. The authors acknowledge Dr Nini Wei at KAUST Core Lab for her assistance in TEM observation.
Notes and references
- G. Cacciola, N. Giordano and G. Restuccia, Int. J. Hydrogen Energy, 1984, 9, 411–419 CrossRef CAS; E. Newson, T. Haueter, P. Hottinger, F. Vonroth, G. Scherer and T. H. Schucan, Int. J. Hydrogen Energy, 1998, 23, 905–909 CrossRef; Y. Okada, E. Sasaki, E. Watanabe, S. Hyodo and H. Nishijima, Int. J. Hydrogen Energy, 2006, 31, 1348–1356 CrossRef PubMed; R. B. Biniwale, S. Rayalu, S. Devotta and M. Ichikawa, Int. J. Hydrogen Energy, 2008, 33, 360–365 CrossRef PubMed; F. Alhumaidan, D. Cresswell and A. Garforth, Energy Fuels, 2011, 25, 4217–4234 CrossRef.
- J. H. Sinfelt, H. Hurwitz and R. A. Shulman, J. Phys. Chem., 1960, 64, 1559–1562 CrossRef CAS; J. H. Sinfelt, J. Catal., 1973, 29, 308–315 CrossRef; J. H. Sinfelt, Science, 1977, 195, 641–646 Search PubMed; J. H. Sinfelt, J. Mol. Catal. A: Chem., 2000, 163, 123–128 CrossRef.
- A. W. Ritchie and A. C. Nixon, Ind. Eng. Chem. Prod. Res. Dev., 1966, 5, 59–63 CrossRef CAS; R. K. Herz, W. D. Gillespie, E. E. Petersen and G. A. Somorjai, J. Catal., 1981, 67, 371–386 CrossRef; L. W. Jossens and E. E. Petersen, J. Catal., 1982, 73, 377–386 CrossRef; A. Touzani, D. Klvana and G. Belanger, Stud. Surf. Sci. Catal., 1984, 19, 357–364 CrossRef; N. Kariya, A. Fukuoka and M. Ichikawa, Appl. Catal., A, 2002, 233, 91–102 CrossRef; S. Hodoshima, H. Arai, S. Takaiwa and Y. Saito, Int. J. Hydrogen Energy, 2003, 28, 1255–1262 CrossRef; S. Hodoshima, H. Nagata and Y. Saito, Appl. Catal., A, 2005, 292, 90–96 CrossRef PubMed; M. Usman, D. Cresswell and A. Garforth, Ind. Eng. Chem. Res., 2012, 51, 158–170 CrossRef; D. Sebastián, C. Alegre, L. Calvillo, M. Pérez, R. Moliner and J. María, Int. J. Hydrogen Energy, 2014, 39, 4109–4115 CrossRef PubMed.
- P. Biloen, J. N. Helle, H. Verbeek, F. M. Dautzenberg and W. M. H. Sachtler, J. Catal., 1980, 63, 112–118 CrossRef CAS; L. W. Jossens and E. E. Petersen, J. Catal., 1982, 76, 265–273 CrossRef; K. Jothlmurugesan, S. Bhatia and R. Srivastava, Ind. Eng. Chem. Fundam., 1985, 24, 433–438 Search PubMed.
- R. B. Biniwale, N. Kariya and M. Ichikawa, Catal. Lett., 2005, 105, 83–87 CrossRef CAS.
- J. W. A. Sachtler and G. A. Somorjai, J. Catal., 1984, 89, 35–43 CrossRef CAS.
- J. Chaouki, D. Klvana and T. Pontier, Chem. Eng. J., 1991, 46, 109–118 CrossRef CAS.
- P. H. Desai and J. T. Richardson, J. Catal., 1986, 98, 392–400 CrossRef CAS.
- J. H. Sinfelt, J. L. Carter and D. J. C. Yates, J. Catal., 1972, 24, 283–296 CrossRef CAS; E. M. Ezzo, H. S. Mazhar, S. A. Ali and N. A. Youssef, Chem. Pap., 1991, 45, 625–641 Search PubMed; S. Yolcular and O. Olgun, Catal. Today, 2008, 138, 198–202 CrossRef PubMed; M. R. Usman, Energy Sources, Part A, 2011, 33, 2231–2238 CrossRef PubMed; S. P. Patil, J. V. Pande and R. B. Biniwale, Int. J. Hydrogen Energy, 2013, 38, 15233–15241 CrossRef PubMed.
- B. Fang, N. K. Chaudhari, M. Kim, J. H. Kim and J.-S. Yu, J. Am. Chem. Soc., 2009, 131, 15330–15338 CrossRef CAS PubMed.
- J. A. Rodriguez and M. Kuhn, J. Chem. Phys., 1995, 102, 4279–4289 CrossRef CAS PubMed; C. Ho, E. Martono, S. Banerjee, J. Roszell, J. Vohs and B. E. Koel, J. Phys. Chem. A, 2013, 117, 11684–11694 CrossRef PubMed.
- G. A. Somorjai, Science, 1985, 227, 902–908 CAS.
- C. H. F. Peden and D. W. Goodman, J. Catal., 1987, 104, 347–358 CrossRef CAS.
- M. R. Usman, D. L. Cresswell and A. A. Garforth, J. Pet. Sci. Technol., 2011, 29, 2247–2257 CrossRef CAS PubMed.
- J. C. Rodríguez, A. J. Marchi, A. Borgna and A. Monzón, J. Catal., 1997, 171, 268–278 CrossRef CAS; R. T. Vang, K. Honkala, S. Dahl, E. K. Vestergaard, J. Schnadt, E. Lægsgaard, B. S. Clausen, J. K. Nørskov and F. Besenbacher, Nat. Mater., 2005, 4, 160–162 CrossRef PubMed; A. K. Rovik, S. K. Klitgaard, S. Dahl, C. H. Christensen and I. Chorkendorff, Appl. Catal., A, 2009, 358, 269–278 CrossRef PubMed; B. Lorenzut, T. Montini, L. De Rogatis, P. Cantonb, A. Benedetti and P. Fornasiero, Appl. Catal., B, 2011, 101, 397–408 CrossRef PubMed; X. Li, C. Zhang, H. Cheng, L. He, W. Lina, Y. Yu and F. Zhao, J. Mol. Catal. A: Chem., 2014, 395, 1–6 CrossRef PubMed.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Science, 2008, 320, 1320–1322 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- S. Bouarab, A. Vega, M. J. López, M. P Iñiguez and J. A. Alonso, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 13279 CrossRef CAS; C. L. Cleveland and U. Landman, J. Chem. Phys., 1991, 94, 7376–7396 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc04016g |
| This journal is © The Royal Society of Chemistry 2015 |